The Setup
In the world of bioinformatics, managing software dependencies and maintaining reproducibility are critical challenges. High-Performance Computing (HPC) environments amplify these challenges, as efficiency, security, and flexibility must be balanced against the need for version control and ease of use. While some bioinformaticians prefer to install software from source for full control, many have adopted modern tools such as Conda, nf-core and containerisation technologies like Apptainer to streamline their workflows and ensure reproducibility.
Recently, I spoke with a colleague who was sceptical of these approaches, preferring to manually install bioinformatics software from source or rely on system admins. They’re not the only ones reluctant to “get with the times” and many I know still - to greater or lesser extent - cling to the old status quo of always installing from source and building pipelines in-house.
But here’s the problem: this approach creates significant portability and reproducibility issues. When you install from source, your workflow is tied to the specific environment where it was compiled. Moving that workflow to another system? You’ll likely face hours of reinstalling software and fixing dependency conflicts. It’s a nightmare for collaboration too. Without strict version control, even tiny software updates can produce variability in your analysis, throwing validation and reproducibility out the window. And that’s a big problem when your research needs to hold-up under peer-review.
My preferred approach, which uses Conda for software installations and nf-core pipelines (powered by Nextflow and Apptainer), tackles these issues head-on. Conda makes sure environments are consistent across machines, resolving dependencies and system-specific conflicts, while nf-core pipelines bundle tools into Apptainer containers, ensuring reproducibility regardless of where the pipeline runs. This method guarantees portability and standardisation, so you can run your analyses anywhere, collaborate easily, and trust that results will be the same every time.
If you’re wondering what Conda is, I wrote a couple of articles on it a while ago which you can link to here.
Doesn’t Compute
A concern often cited by the old-school, and one reason they will still prefer to install software manually, is that containerisation technologies may introduce performance overhead compared to running software natively. On HPC systems, where every ounce of computational efficiency counts, adding another layer of abstraction seems counterintuitive. However, much of this scepticism stems from experiences with older virtualisation technologies, which did indeed slow things down, but modern solutions such as Apptainer (formerly Singularity) are built specifically for HPC, integrating directly with the system kernel for near-native performance.
Another concern is that containers might not fully support specialised HPC hardware like GPUs or high-speed interconnects (e.g., InfiniBand). While early container tech struggled with these configurations, Apptainer is designed to handle HPC-specific setups, offering direct access to hardware and interconnects without compromising performance.
Some also prefer to install from source for more granular control. Customising the compilation process and fine-tuning dependencies offers full control over the environment. But containers abstract this away, bundling everything into a single image. For those accustomed to managing every detail, this can feel restrictive.
Finally, there’s the issue of security. In shared computing environments, security is a priority, and tools like Docker require root privileges, which can introduce risks. Apptainer, on the other hand, was built with security in mind for multi-user HPC systems. It doesn’t require root access, making it a much safer option for containerised workflows.
Container Confidence
Installing Nextflow and the nf-core pipelines using Conda is now the approach recommended by nf-core (though I am convinced they nicked this from me). Conda resolves dependencies, isolates environments, and ensures changes stay contained. With version control and rollback features, it’s easy to switch between different versions of Nextflow or nf-core tools.
One of the greatest advantages of nf-core pipelines is their use of Apptainer containers for version control and reproducibility. On HPC systems, Apptainer is installed system-wide by admins for security reasons. As mentioned, Apptainer doesn’t require root access, making it safer than alternative tools like Docker in shared environments. This setup means I can simply load the module in my driver job script for seamless, secure execution.
Each nf-core pipeline is tied to a specific set of containers bundling versions of the tools from which it is comprised. As shown in the schematic below, Nextflow automatically pulls the pipeline from GitHub and fetches the correct containers for each tool in the pipeline. This ensures that every run, no matter where it’s executed, uses the same software environment. Results are easy to replicate because the environment stays consistent from run to run, which is crucial for reproducibility.
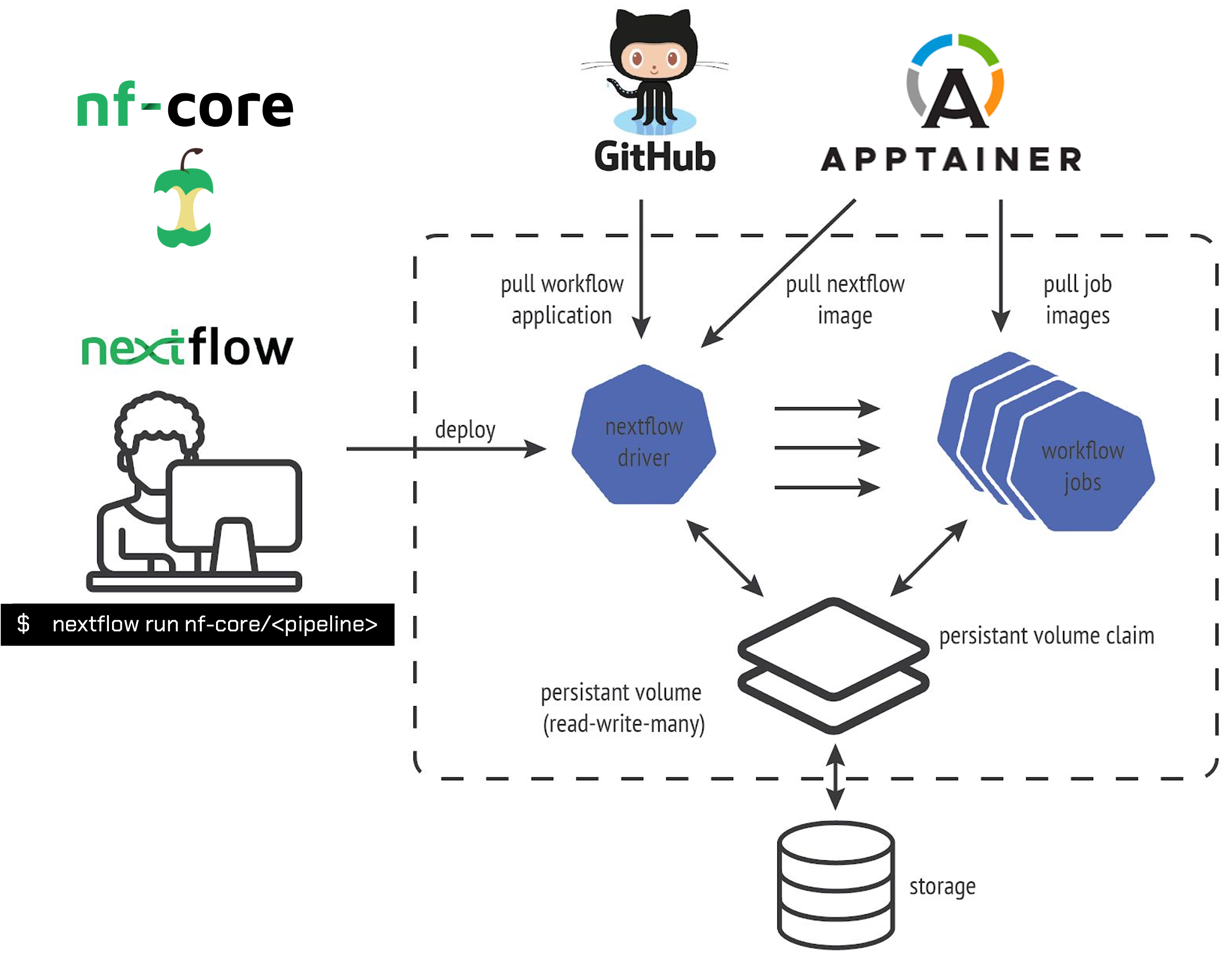
While standardised nf-core pipelines handle most of my high-throughput workflow needs, there are times when I need to run more bespoke analyses. For example, I might need to work with bioinformatics tools or run analyses that are not yet part of nf-core. In these cases, I turn to Conda to create specialised environments containing the tools required to run my custom scripts, both versioned with Git and made available through GitHub.
Conda is particularly useful for this kind of flexibility because it allows me to quickly set up new environments and install tools without worrying about container images. I can easily switch between different environments depending on the project, keeping my analyses isolated and reducing the risk of software conflicts.
Building containers for preservation and shareability then offers a long-term solution for ensuring reproducibility and portability. Containers encapsulate the entire environment, allowing others to easily reproduce the exact same conditions, making collaboration smoother and ensuring future-proofing for complex bioinformatics projects.
In summary, this hybrid approach using Conda for managing installations and nf-core pipelines underpinned by containerisation offers several clear advantages over historical approaches:
1. Reproducibility
With nf-core pipelines using containers, every analysis is fully reproducible. Containers lock down software versions, ensuring consistent results across systems and timeframes; crucial in bioinformatics, where tools and dependencies evolve rapidly.
2. Version Control
Conda and nf-core together provide robust version control. Conda allows me to manage and update the versions of Nextflow and nf-core tools, while the pipelines themselves automatically pull the correct containers for the analysis depending on the version of the pipeline that is specified. This dual-layered versioning ensures that I can control the software at both the pipeline and environment levels.
3. Security and Performance
Apptainer is optimised for HPC environments and offers near-native performance, mitigating concerns about container overhead. Furthermore, by running without root privileges, Apptainer enhances the security of the system, making it a safer option than Docker for shared computing resources.
4. Flexibility
While containers provide stability and reproducibility, Conda offers the flexibility to run bespoke or experimental analyses. I can set up custom environments for specialised workflows without needing to build new container images or worry about container configuration.
5. Ease of Use
Managing software installations through Conda is straightforward, and nf-core pipelines further simplify bioinformatics workflows by automating the management of containers and dependencies. This means I spend less time resolving software conflicts and more time focusing on the actual analysis.
Conclusion
By combining Conda for software management with nf-core pipelines underpinned by containerisation for reproducibility and security, I believe that I have leveraged the best of modern bioinformatics tools to create a workflow that balances flexibility and control.
For bioinformaticians working in HPC environments, this hybrid model offers a modern, scalable solution, combining containerisation’s strengths with the control and performance required for complex analyses. It solves challenges of dependency management, reproducibility, and security, while remaining flexible enough to adapt to new tools and workflows.
For those still in doubt - how are those source installs treating your productivity?
Thanks for reading. I hope you enjoyed the article and that it helps you to get a job done more quickly or inspires you to further your data science journey. Please do let me know if there’s anything you want me to cover in future posts.
If this tutorials has helped you, consider supporting the blog on Ko-fi!
Happy Data Analysis!
Disclaimer: All views expressed on this site are exclusively my own and do not represent the opinions of any entity whatsoever with which I have been, am now or will be affiliated.